アストロサイトに関するプロジェクト
ヒトを含む哺乳類の脳、脊髄には、ニューロンとグリア(アストロサイト 、オリゴデンドロサイト、ミクログリア)と呼ばれる細胞が存在します。これまで、脳や脊髄の働きは、主にニューロンによって担われ、グリア細胞は単にニューロンをサポートするための細胞と考えられてきました。このため、脳卒中や脊髄損傷ではニューロンがダメージを受け、主にニューロンの機能異常が運動麻痺などの病態の原因であると考えられてきました。しかし、最近の研究により、グリア細胞の一つであるアストロサイトも、ニューロン同様、脳、脊髄の機能において重要な役割を果たすことが明らかにされつつあり、アストロサイトの機能異常もその病態に深く関与することが示唆されています。アストロサイトは発生期に、ニューロンと同様に神経幹細胞から産み出されます。アストロサイト産生の分子メカニズムは不明な点も多く、現在も研究が続けられています。また、アストロサイトは脳卒中や脊髄損傷などの損傷時に反応性アストロサイトとなり、炎症反応、グリア瘢痕の形成、神経保護など、機能回復にとって良い働きも悪い働きもすることが知られています。私たちの研究室では、これらのアストロサイトの働きを担う分子メカニズムを明らかにし、うまくコントロールすることで損傷した中枢神経系の再生・修復が可能かを調べています。
アストロサイト産生に関わる新しい転写因子Zbtb20の発見
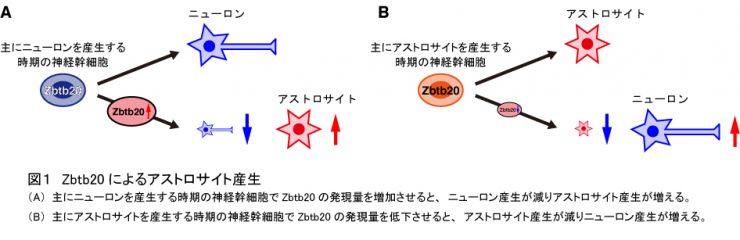
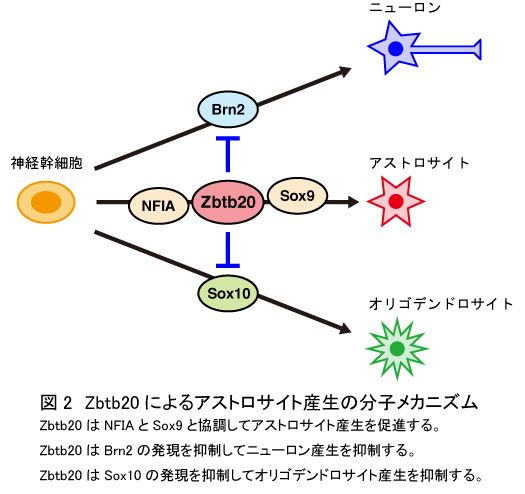
脳、脊髄などの中枢神経系の発生過程において、神経幹細胞は、先にニューロンを産生し、その後、アストロサイトを産生します。神経幹細胞がアストロサイトを産生し始める時期に、神経幹細胞で発現量が高くなる因子を探索し、転写因子Zbtb20を発見しました。主にニューロンを産生する時期の神経幹細胞において、Zbtb20の発現量を増加させたところ、ニューロン産生量が減り、神経幹細胞は主にアストロサイトを産生するようになりました(図1A)。一方、アストロサイトを産生する時期の神経幹細胞において、Zbtb20の発現量を低下させたところ、アストロサイト産生量は減り、代わりに神経幹細胞はニューロンを産生するようになりました(図1B)。以上の結果は、Zbtb20が神経幹細胞で発現し働くと、神経幹細胞はアストロサイトを産み出すようになることを示しています。次に、私たちは、Zbtb20がどのようなメカニズムでアストロサイト産生を制御するのかを調べました。グリア細胞(アストロサイトとオリゴデンドロサイト)産生を制御する因子としてSox9、NFIAと呼ばれる転写因子が知られていました。今回の研究により、Zbtb20はSox9とNFIAの2つの因子と協調して働き、アストロサイト産生を促進していることが明らかになりました。さらに、Zbtb20はニューロン産生に必須の転写因子Brn2、オリゴデンドロサイト産生に必須の転写因子Sox10の発現を抑制し、神経幹細胞がニューロンやオリゴデンドロサイトを産生するのを抑えていることが示されました。このようなメカニズムによって、Zbtb20はアストロサイト産生を制御していると考えられます(図2)。この成果はNature Communications誌に発表されました(Nagao et al, Nat Commun 7, 11102, 2016)。
クロマチン制御因子HMGNファミリータンパク質によるアストロサイト分化の促進
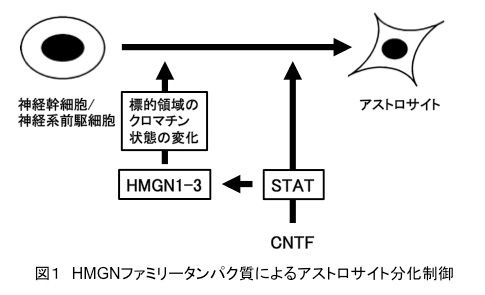
アストロサイトは、ニューロンへの栄養供給、ニューロンが放出した神経伝達物質の回収、シナプス周辺のイオン環境の維持、血液脳関門の制御、シナプス形成など、中枢神経系の様々な機能において重要な役割を果たします。アストロサイトはニューロン、オリゴデンドロサイトと同様に神経幹細胞から産み出されます。近年の研究により、STATシグナル経路がアストロサイト分化を促進することはよく知られていますが、アストロサイト分化の分子メカニズムについては未だ不明な点も多く残されています。私たちは、脳の発生過程において、クロマチン制御因子であるHMGNファミリータンパク質(HMGN1、2、3)が、アストロサイト分化を促進することを明らかにしました。培養したマウス神経系前駆細胞で、HMGN1、2、3を過剰発現させたところ、アストロサイト分化は促進され、逆に、ノックダウンすると、アストロサイト分化の抑制が観察されました。さらに、マウス個体内の神経系前駆細胞でHMGN1、2、3を過剰発現またはノックダウンしても、同様の結果が得られました。これらの結果は、HMGNファミリータンパク質が神経幹細胞のアストロサイトへの分化を促進することを示しています。重要なことに、HMGNファミリータンパク質の過剰発現はSTATを活性化しませんが、HMGNファミリータンパク質のノックダウンは、STATシグナル経路を活性化する毛様体神経栄養因子(Ciliary neurotrophic factor: CNTF)によるアストロサイト分化促進を顕著に抑制しました。以上より、HMGNファミリータンパク質は、STATシグナル経路と並列またはその下流ではたらき、アストロサイト分化を促進する新規のクロマチン制御因子であることが示唆されました(図1)。この成果はStem Cells誌に発表されました(Nagao et al, Stem Cells 32, 2983-2997, 2014)。
オリゴデンドロサイトに関するプロジェクト
オリゴデンドロサイトはニューロンに巻き付くことで髄鞘を形成し、活動電位伝播の促進と軸索の安定性に寄与しています。脊髄損傷や脱髄変性疾患(多発性硬化症など)では髄鞘が破壊され、ニューロンの細胞死や神経伝達の障害が起こり、四肢の麻痺や痙縮を引き起こします。脊髄損傷や脱髄変性疾患において症状を改善させるためには、この破壊された髄鞘を再生させる(再髄鞘化)必要があります。現在のところ、再髄鞘化を促進する有効な治療法はなく、新しい治療法開発のための研究が行われています。私たちの研究室では、再髄鞘化に関わる新しい分子の探索を行っています。
脊髄損傷後の再髄鞘化に関わる新しいクロマチン制御因子Chd7の発見
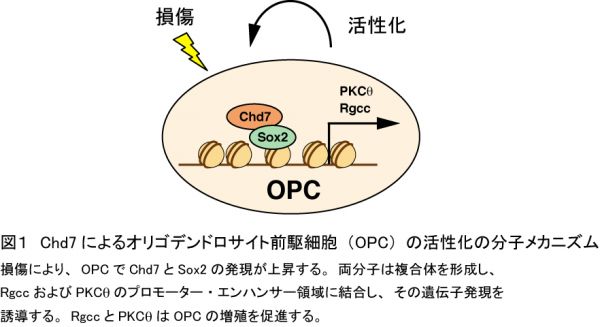
脊髄損傷後の再髄鞘化促進は、損傷脊髄の機能回復に向けた重要な治療戦略のひとつと考えられています。再髄鞘化を促進するためには、オリゴデンドロサイト前駆細胞(OPC)を活性化し、その増殖、分化、成熟を促進することが必要ですが、この過程の分子メカニズムは未だ不明な点が多く残されています。私たちは、クロマチン制御因子Chd7が脊髄損傷後のOPCの活性化を制御することを明らかにしました。Chd7は成体脊髄において、一部のOPCとオリゴデンドロサイトで発現し、脊髄損傷後、増殖するOPCでその発現は顕著に増加することがわかりました。このChd7の発現パターンから、Chd7はOPCの活性化に関与する可能性が考えられ、OPC特異的Chd7欠損マウスを用いてその可能性を検討しました。このマウスでは、脊髄損傷後、OPCの増殖、オリゴデンドロサイトへの分化および再髄鞘化が顕著に抑制されていました。また、BMSスコアによる後肢運動機能回復評価を行ったところ、このマウスでは、コントロールと比較して有意にその回復が悪いことがわかりました。これらの結果から、Chd7はOPCの増殖、オリゴデンドロサイトへの分化、その後の再髄鞘化に必要であることが示唆されました。次に、OPCの培養系を用いて、Chd7がどのようにOPCを活性化するのか、その分子メカニズムを調べました。その結果、Chd7は転写因子Sox2と複合体を形成し、増殖に関与する新規分子RgccおよびPKCθの発現を直接誘導しOPCを活性化することが明らかになりました(図1)。この研究成果はJournal of Neuroscience誌に発表されました(Doi et al, J Neurosci 37, 10290-10309, 2017)。